
by Soultana Tatsika, MSc Student in Bioeconomy / International Hellenic University
The advances in DNA sequencing, with the development of Next Generation Sequencing (NGS) allow the comprehensive examination of microbial communities, without the need of cultivation (Hall 2007). Thus, instead of examining the genome of an individual bacteria strain that has been grown in a laboratory, methods, based on gene fragment DNA sequencing, gives the advantage of analysing the genetic material coming directly from microbial communities sampled from natural environments (figure 1). Millions of DNA sequences can be determined simultaneously in multiple samples without cloning, due to the fact that high – throughput sequencing (HTS) approaches are based on the principle of sequencing – by – synthesis and the strategy of massively parallel signature. These methods are offering high specificity and sensitivity in an relative inexpensive way while being less time consuming comparing with traditional molecular methods for microbial diversity research (Nucleic Acid Hybridization Based methods, PCR Electrophoresis based methods and Sager Sequencing based methods) (Xu W., 2016). It is now clear that these methods offer the opportunity of an in – depth study of the microbial diversity in food which can be achieved after direct nucleic acid extraction from the sample studied (Ercolini 2013). The development of these NGS technologies and their application in the field of food ecosystems showed that these communities are perhaps more rich than expected (Kergourlay 2015).
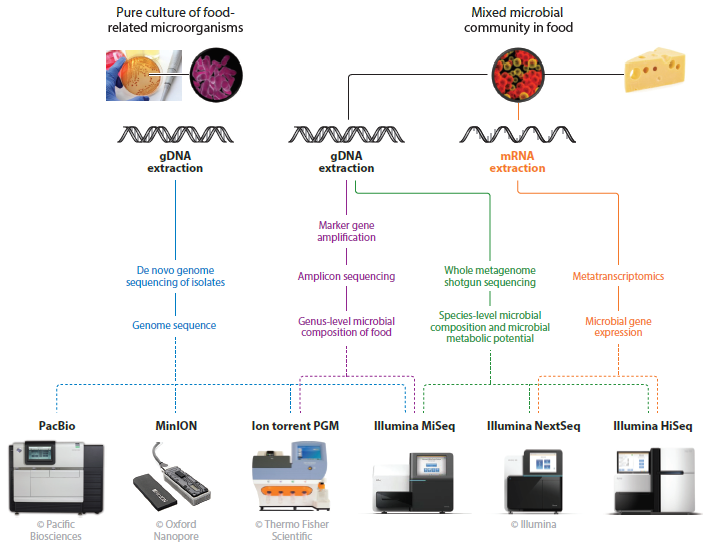
Figure 1. Overview of the different HTS sequencing approaches applicable to food microbiology and recommendations for the sequencing platforms which are most suitable for each approach (Walsh et al, 2017).
In HTS applications, two substantially different approaches are used: targeted amplicon sequencing and shotgun sequencing (figure 2). Generally, in amplicon sequencing, a phylogenetically informative marker is PCR amplified from DNA extracted from a mixed microbial community and sequenced and aligned against a reference database to determine the taxonomic composition of a sample. The most commonly used amplicon sequencing method is 16S rRNA gene sequencing, which is used to profile bacterial communities (Claesson et al., 2010; Liu et al 2007; Ronaghi and Elahi 2002; Walsh et al. 2017). In shotgun metagenomics, direct sequencing of bacterial DNA extracted from a microbial community is used and the sequencing reads are analyzed using metagenomic databases as a reference, targeting to the assignment of each sequencing read to a specific taxon (Truong et al., 2015). In the shotgun based approach, the whole genomic potential (metagenome) of the microbial populations in a given sample can be determined. It is reported that shotgun metagenomics permits in general much deeper characterization of the microbiome complexity, allowing identification of a greater number of species per sample, compared to 16S rRNA gene amplicon sequencing (Laudadio et al., 2018). When mRNA is the target of the analysis (after cDNA synthesis) the whole pool of genes actually expressed in food sample (metatranscriptome) can be determined, providing basic information of how the microbial communities interact to each other (Franzosa Ea., et al, 2015). The result is a mixed expression profile which characterizes the expression behaviour of entire microbial communities in response to different conditions.

Figure 2: Application of HTS sequencing for the study of food microbiota or microbiome (De Filippis F., et al, 2018). Abbreviations: NA, nucleic acid; OUT, operational taxonomic unit; PCR, polymerase chain reaction.
16S ribosomal (rRNA) gene sequence is a conserved phylogenetic marker which can be reliably amplified by a range of “universal primers” targeted at its conserved regions (Pace, 2009; Pace et al., 1986). It is made up of nine hypervariable regions flanked by conserved sequences (figure 3)(Neefs et al., 1993). By amplifying and sequencing these hypervariable loci, identification of the corresponding bacterial taxonomy of the species associated with sample can be achieved (Cox et al., 2013). rRNA amplicons obtained from DNA/RNA extracted directly from food matrixes, are sequenced and the sequences are compared to reference databases in order to identify the Operational Taxonomic Units (OTUs) through existing bioinformatics pipelines. OTUs – a term used to describe closely related individuals – are determined based on sequence similarity, where a percentage of >97% similarity, constitutes the same OTU (Schloss et al., 2011). A level of 97% of sequence identity is usually preferred as being representative of a species and a level of 95% for a genus when partial 16S rRNA gene sequences are used (Cox et al., 2013). The approach is qualitative but it can also be considered as semi – quantitative due to the fact that the number of sequence reads identified with the same OTU permits an estimation of the relative abundance of each bacterial entity in the food matrix analyzed (Cocolin and Ercolini, 2015).
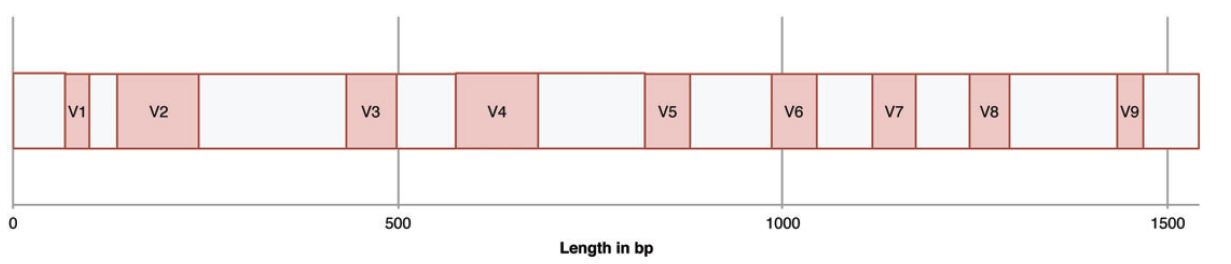
Figure 3: 16S rRNA gene (approximately 1,540kb in length) of E. coli showing the nine variable loci which allow the discrimination between different microorganisms (Cox et al., 2013).
From the generated sequences in NGS analysis, information can be obtained about the microbial taxonomy of culturable and non culturable bacteria in a food sample and theoretically information on the presence of undesirable microorganisms (e.g. foodborne pathogens). Recently, there is an increasing opportunity for its application due to the fact that the price of NGS platforms and their running costs are decreasing (Lecruit and Eloit, 2014). However, detection of specific bacterial species remains difficult due to issues associated with specificity and sensitivity, for example false positive or negative detections of sequences shared between different bacterial species (Borthong J 2018). 16S rRNA metagenomic analysis uses PCR, thus the results can be affected by this amplification step. The selection of primers used to amplify rRNA amplicon is crucial because some primers have been shown to display a bias leading to over- or under- representation of specific taxa (Rintala et al, 2017). Problems also may arise, due to chimeric sequences in PCR products (Haas et al., 2011) and closely related bacteria cannot be differentiated (Weinstock, 2012). Finally, there are interpretation issues arising when using these methods to food quality control such as weaknesses in differentiation of viable and non viable microorganisms in the sample, due to the fact that nucleic acids have been shown to persist after inactivation of microorganisms (Ceuppens S. et al., 2014). Nevertheless, for the characterization of food related microbiota, 16S rRNA gene sequencing is considered one of the most popular high – throughput sequencing method, mainly due to the availability of many easy to operate and free for use bioinformatic tools, designed for sequencing data analysis (Cao et al., 2017). The most popular software to analyze sequencing data from food matrixes are QIIME (Quantitative Insights Into Microbial Ecology) (Caparaso et al, 2010), mothur (Schloss et al, 2009) and USEARCH (ultra – fast sequence analysis) (Edgar, 2010).
Recently, a metagenome predictive tool, called PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States_http://picrust.github.io/ picrust), has been developed, in order to predict the functional composition of a bacterial community present in a food sample using marker gene data (16S rRNA gene amplicon data) and a database of reference genomes (Langille et al, 2013). This new area of research is called Predictive Metagenomic Profiling (PNP) and only requires sufficient sequencing depth to cover a single target gene reducing the amount of data needed for a preliminary depiction of the community functional profile (Howe et al., 2014). PMP pipeline produces a table containing the predicted gene family counts as KEGG ontology identifiers which can be grouped to path – way categories like KO modules. Moreover, for each OTU, 16S rRNA gene amplicon copy number can be used to measure the relative contribution of predicted gene families to the community’s functional potential (Wood, 2016). The results of PMP pipeline can be used to support Microbial Risk Assessment (MRA) studies concerning the prediction of a phenotypic behaviour like antimicrobial resistance of the identified OTU concerned (Cocolin et al., 2017). PMP analysis is considered as a potential link between metagenomics and MRA (Figure 3).

Figure 4: Potential link between NGS and MRA (Cocolin et al., 2017). Abbreviations: NGS, Next Generation Sequencing; MRA, Microbial Risk Assessment.
Most of the food microbiome related NGS studies have focussed on fermented foods like cheese, Kimchi (naturally fermented from vegetables), Kefir (a symbiotic community – fermented beverage), fermented seafood and sausages (Kergourlay et al., 2015; De Filippis et al., 2017; Patra et al., 2016). Food fermentation as a dynamic process involves continuous alterations of microbial communities’ composition and amplicon sequencing has been used to characterize these changes, offering information for the identification of biomarkers for the ripeness and quality or spoilage of fermented food (De Pasquale et al., 2014; Polka et al., 2015; Walsh el al., 2016). These tools have been also applied to the study of microbial communities of fresh and non – fermented foods like fresh fruit and vegetables, raw and pasteurised milk, poultry etc (Leff and Fierer, 2013; Quigley et al., 2013; Oakley et al., 2013;). For non – fermented foods, these new tools can offer non – targeted perception of food microbial community conditions post – harvest and post – processing, with implications for food safety and stability (Bokulich et al, 2016). Finally, there are studies using amplicon – sequencing determination of the microbial taxa present in food processing facilities, with the meat and dairy processing environments to be the most commonly studied ones (Doyle et al., 2017).
Acknowledgments
We are grateful to Ms Tatsika for kindly providing the original article.
References
- Bokulich, N.A., Lewis Z.T., Boundy – Mills, K., and Mills D.A. (2016). A new prespective on microbial landscapes within food production. Curr Opin Biotechnology. Vol 37: 182-189. DOI:10.1016/j.copbio.2015.12.008.
- Borthong J, Omori R, Sugimoto C, Suthienkul O, Nakao R and Ito K (2018). Comparison of Database Search Methods for the Detection of Legionella pneumophila in Water Samples Using Metagenomic Analysis. Front. Microbiol. 9:1272. doi: 10.3389/fmicb.2018.01272
- Cao Yu, Seamus F., Sinead P., Kieran J. and Shabarinath S. (2017), A Review on the Application of Next Generation Sequencing Technologies as Applied to Food Related Microbiome Studies. Frontiers in Microbiology, Vol 8, article 1829, pp.1-16.
- Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
- Ceuppens, S., Li M., Uyttendaele, M., Renault, P., Ross, P., Van Ranst, M., Cocolin, L. and Donaghy J. (2014). Molecular Methods in Food Safety Microbiology: Interpretation and Implications of Nucleic Acid Detection. Comprehensive Reviews in Food Science and Food Safety, Vol 13: 551-574. doi: 10.1111/1541-4337.12072
- Claesson MJ, Wang Q, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O’Toole PW. (2010). Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38: 200.
- Cocolin, L, and Ercolini, D., (2015). Zooming into food – associated microbial consortia: a “cultural” evolution. Current Opinion in Food Science, Vol 2: 43-50.
- Cocolin, L., Mataragas, M., Bourdichon, F., Doulgeraki, A.,, Pilet, M.F., Jagadeesan, B., Rantsiou, K., Phister, T. (2017). Next generation microbiological risk assessment meta-omics: The next need for integration. Int J Food Microbiol. pii: S0168-1605(17)30502-0. doi: 10.1016/j.ijfoodmicro.2017.11.008
- Cox M.J., Cookson W.O.C.M. and Moffatt M.F. (2013). Sequencing the human microbiome in health and disease. Human Molecular Genetics Vol. 22, Review Issue 1. DOI:10.1093/hmg/ddt398
- De Filippis F., Parente E. and Ercolini D. (2018). Recent Past, Present and Future of the Food Microbiome. Annual Review of Food Science and Technology, 9:25.1 – 25.20. https//doi.org/10.1146/annurev-foof-030117-012312.
- De Pasquale I, Di Cagno R, Buchin S, De Angelis M, Gobbetti M. 2014. Microbial ecology dynamics reveal a succession in the core microbiota involved in the ripening of pasta filata caciocavallo pugliese cheese. Appl. Environ. Microbiol. 80:6243–55.
- Doyle, C.J., O’ Toole, P.W., and Cotter, P.D. (2017). Metagenome-based surveillance and diagnostic approaches to studying the microbial ecology of food production and processing environments. Environmental Microbiology, 19(11): 4382-4391. DOI:10.1111/1462-2920.13859.
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
- Ercolini, D. (2013). High-throughput sequencing and metagenomics: moving forward in the culture-independent analysis of food microbial ecology, Appl. Environ. Microbiol. 79. pp. 3148-3155.
- Franzosa, Ea, Hsu, T., Sirota-Madi, A., et al. Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nat Rev Microbiol. 2015; 13:360–372. [PubMed: 25915636] [An overview of microbial community profiling techniques and their integration with ‘omics’ data.]
- Haas, B. J., Gevers, D., Earl, A. M., Feldgarden, M., Ward, D. V., Giannoukos, G., et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504. doi: 10. 1101/gr.112730.110
- Hall, N. (2007). Advanced sequencing technologies and their wider impact in microbiology. J. Exp. Biol, 210, pp. 1518 – 1525.
- Howe, A.C., Jansson, J.K., Malfatti, S.A., Tringe, S.G., Tiedje, J.M., Brown, C.T., 2014. Tackling soil diversity with the assembly of large, complex metagenomes. Proceedings of the National Academy of Sciences of the United States of America, 111, 4904-4909.
- Kergourlay, G., Taminiau, B., Daube, G., Champomier, Verges M.C. (2015). Metagenomic insights into the dynamics of microbial communities in food, Int. J. Food Microbiol. 213, pp 31-39.
- Langille, M.G.I., Zaneveld, J., Caporaso, J.G., McDonald, D., Knights, D., Reyes, J.A., Clemente, J.C., Burkepile, D.E., Vega Thurber, R.L., Knight, R., Beiko, R.G., Huttenhower, C., 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814-821.
- Laudadio, I., Fulci, V., Palone, F., Stronati, L., Cucchiara, S. and Carissimi, C. (2018). Quantitative Assessment of Shotgun Metagenomics and 16S rDNA Amplicon Sequencing in the Study of Human Gut Microbiome. OMICS A Journal of Integrative Biology. Vol 22, Number 4. DOI:10.1089/omi.2018.0013.
- Lecuit, M., and Eloit, M. (2014). The diagnosis of infectious diseases by whole genome next generation sequencing: a new era is opening. Front. Cell. Infect. Microbiol. 4:25. doi: 10.3389/fcimb.2014.00025
- Leff, J.W. and Fierer, N. (2013). Bacterial Communities Associated with the Surfaces of Fresh Fruits and Vegetables. Plos ONE 8 (3): e59310. doi:10.1371/journal.pone.0059310.
- Liu, Z., Lozupone, C., Hamady, M., Bushman, F.D. and Knight, R. (2007) Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35, 120.
- Neefs, J. M., Van de Peer, Y., De Rijk, P., Chapelle, S., and De Wachter, R. (1993). Compilation of small ribosomal subunit RNA structures. Nucleic Acids Res. 21, 3025–3049. doi: 10.1093/nar/21.13.3025
- Oakley BB, Morales CA, Line J, et al. (2013). The poultry-associated microbiome: network analysis and farm-to-fork characterizations. PLOS ONE.; 8:e57190. [PubMed: 23468931]
- Pace NR (2009) Mapping the tree of life: progress and prospects. Microbiol Mol Biol Rev 73(4):565–576
- Pace NR, Olsen GJ et al (1986) Ribosomal RNA phylogeny and the primary lines of evolutionary descent. Cell 45(3):325–326.
- Patra, J. K., Das, G., Paramithiotis, S., and Shin, H.-S. (2016). Kimchi and other widely consumed traditional fermented foods of Korea: a review. Front. Microbiol. 7:1493. doi: 10.3389/fmicb.2016.01493
- Połka J, Rebecchi A, Pisacane V, Morelli L, Puglisi E. (2015). Bacterial diversity in typical Italian salami at different ripening stages as revealed by high-throughput sequencing of 16S rRNA amplicons. Food Microbiol. 46:342–56
- Quigley L, McCarthy R, O’Sullivan O, et al. (2013). The microbial content of raw and pasteurized cow milk as determined by molecular approaches. J Dairy Sci.; 96:4928–4937. [PubMed: 23746589]
- Rintala A, Pietila¨ S, Munukka E, et al. (2017). Gut microbiota analysis results are highly dependent on the 16S rRNA gene target region, whereas the impact of DNA extraction is minor. J Biomol Tech 28, 19–30.
- Ronaghi, M. and Elahi, E. (2002) Pyrosequencing for microbial typing. J Chromatogr B Analyt Technol Biomed Life Sci 782, 67–72.
- Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., Lesniewski, R.A., Oakley, B.B., Parks, D.H., Robinson, C.J. (2009). Introducing mother: Open – source, platform – independent, community – supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23),7537-7541. doi:10.1128/AEM.01541-09.
- Schloss, P.D., Gevers, D., and Westcott S.L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies, PLoS One 6:e27310. http://dx.doi.org/10.1371/journal.pone.0027310.
- Truong DT, Franzosa EA, Tickle TL, et al. (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 12, 902–903. DOI: 10.1038/nmeth.3589
- Walsh, M.A., Crispie F., Claesson J.M, Cotter D.P. (2017). Translating Omics to Food Microbiology. Annual Review Food Sci. Technology. 8:6.1-6.22
- Weinstock, G. M. (2012). Genomic approaches to studying the human microbiota. Nature 489, 250–256. doi: 10.1038/nature11553
- Wood, J., 2016. Predictive metagenomics profiling: why, what and how? Bioinformatics Rev. 2, 1–4.
- Xu W. (2016) Characterization of Microbial Diversity in Food Chain: A Molecular Review in Functional Nucleic Acids Detection in Food Safety, Chapter 16. Springer Science + Business Media Singapore. DOI:10.1007/978-981-10-1618-9_16.